魏星 , 易敏寒
, 易敏寒
Wei Xing, Yi Minhan
中图分类号: TP391 G202
通讯作者:
收稿日期: 2017-07-3
修回日期: 2017-07-28
网络出版日期: 2017-10-25
版权声明: 2017 《数据分析与知识发现》编辑部 《数据分析与知识发现》编辑部
基金资助:
展开
摘要
【目的】在海量文献中, 挖掘并预测生物医学实体之间的新关联, 构建关联网络。【方法】提出一种基于数据立方体的新方法挖掘疾病-基因-药物间关联, 以糖尿病为例, 构建关联网络, 并使用关联规则量化实体关联程度。【结果】由糖尿病相关疾病(14种)、基因(23种)和药物(24种)构建三个1-D方体、三个2-D方体及其关联网络和一个3-D方体关联网络, 共计存在411种关联, 同时得到8个关联子网。【局限】数据预处理存在主观性, 可能会对挖掘结果产生影响。【结论】算法性能优于其他同类算法, 能够为糖尿病精准医疗提供更好的新研究思路。
关键词:
Abstract
[Objective] This study aims to construct a disease-gene-drug correlation network for diabetes mellitus (DM). [Methods] First, we proposed a new data cube-based approach to construct a disease-gene-drug correlations network for the DM. Then, we measured the associations among the biological entities. [Results] We retrieved the needed data from the PubMed database and constructed three 1-D vertex cubes, three 2-D square cubes and one 3-D disease-gene-drug network, which revealed 411 associations among the 14 subclasses of DM, 23 genes, and 24 drugs. We also constructed 8 optimal disease-gene-drug subnetworks of DM. [Limitations] There were some subjective issues with the data analysis. The changing of user behaviors may also influence the results. [Conclusions] The proposed algorithm is better than the existing ones, which provides new directions for research on customized medical treatments.
Keywords:
生物医学文献正在以前所未有的速度增长, 其摘要中包含了海量的实验结果、基因表型描述和药效信息, 整理挖掘其中有效信息, 已成为生物知识发现和生物医学研究中一个重要手段[1]。如何才能有效利用这些文本中所蕴含的生物医学知识, 无疑对分析海量生物医学数据是非常重要的, 常用方法是通过关键词直接检索, 但是这只能从大量文档集合中找到用户需求相关的文件列表, 而不能从文本中直接获取用户感兴趣的信息。因此, 如何从大规模生物医学文献中自动挖掘相关知识是一项迫在眉睫的任务。常见的生物实体间关联的研究有: 蛋白质与基因的关联[2], 药物与药物的关联[3], 药物与疾病的关联[4]等。
数据立方体(Data Cube)[5]能够存放多个数据维(如疾病、基因和药物)上的预计算度量(如关联强度), 用户可以以多维方式, 通过如下钻或上卷这样的联机分析处理(OLAP)操作探查数据, 进行数据分析和知识发现, 探索感兴趣的模式。
本文基于数据立方体探查多维空间中的数据, 同时使用关联规则计算实体间的关联度, 以糖尿病为例, 构建糖尿病相关疾病-基因-药物关联网络, 分析并探讨实体间潜在关联, 突出并挖掘关联网络中的关键节点, 提出实验性研究假设, 为研究人员对今后有关糖尿病的诊断与治疗、疾病候选基因筛选、靶向药物和个性化医疗等研究提供数据支持和新的研究思路。
目前与疾病有关的生物医学文本挖掘研究大多集中在基因的功能信息上, 如: 对疾病基因和疾病候选基因的分类排序[6], 使用图论构建疾病与疾病基因关联度的网络模型[7], 利用定量性框架模型综合分析疾病基因与蛋白质之间的作用预测药物新靶点[8]以及计算药物重新定位[9]等, 而关于疾病与多个其他实体的关联挖掘属于一个新兴的研究领域。
生物实体关联挖掘方法有多种, 如: Lamb等[10]利用具有生物活性的小分子治疗基因表达谱数据, 开发“Connectivity Map”系统, 用于挖掘化学与生理过程、疾病与药物之间的小分子共享作用机理, 依此挖掘疾病-药物之间的关联。Natarajan[11]在文献中获得疾病、基因的多种特征, 从OMIM得到已知疾病-基因关联, 对比之后, 挖掘出120对基因-疾病新关联。Odibat等[12]提出一种基于排序任意重叠定位协同聚类算法, 并依此构建判别模型, 通过对基因表达数据集的分析运算, 可以有效分类疾病基因表达结果。Li等[13]构建了一个用于判断疾病与候选基因随机集优先级的评分模型, 使用基于网络与表型分析的方法在生物医学文献中进行数据挖掘, 该模型能够较为精准地将已知致病基因进行排序, 同时也能在一定程度上预测新的候选疾病基因。这些研究使用不同方法挖掘生物实体关联, 为相关研究提供了多种思路, 但使用数据立方体挖掘三个生物实体关联的方法, 笔者所知, 尚未见报道。CoPub[14]和PubGene[15]在两者关联挖掘中与本文方法较为类似, 但前者挖掘了基因-疾病、药物-疾病的关联, 其结果经ROC曲线验证后, 最高只有70% (R-scaled Score≥20), 而后者只挖掘了基因-基因间的关联, 结果精确度仅有60%, 而且这两项研究并没有将三者关联综合构建网络, 分析不够全面。
综上, 目前大多数关联挖掘方法都是在两个生物实体之间进行研究的, 对三个及三个以上的实体关联挖掘方法研究较少, 而且结果精度均不高, 这对预测结果的可信度会造成较大影响。因此, 本文基于数据立方体将疾病-基因-药物三者结合构建关联网络, 挖掘三者之间的新关联, 提高算法性能以及挖掘精确度。
疾病基因药物数据立方体关联网络是由两两关联组合构成的, 实现步骤如下:
(1) 对文献进行数据预处理, 获得数据立方体的0-D顶点方体和三个1-D方体;
(2) 设定最小support阈值, 依据关联规则计算得到三个2-D方体内疾病、基因和药物之间的两两关联度;
(3) 使用BUC算法构建数据立方体, 得到3-D基本方体内的实体间关联度;
(4) 利用R语言实现多维方体的关联网络的可视化, 分析关联网络的分布程度和不同模式的识别程度。
(5) 使用ROC曲线验证本文算法的准确性和可靠性。
由于文献摘要是自然语言书写, 属于非结构化数据, 所以需先进行数据标准化预处理, 不同研究者侧重点不同, 本文设定如下步骤进行处理:
(1) 将文献摘要所有字母转为小写;
(2) 把文本转化为单独句子;
(3) 去除标点符号以及与本研究无关的词, 如: “this”、“an”等;
(4) 替换希腊字母, 如: “α→Alpha”等;
(5) 基于词典(Gene_Dictionary”和“Drug_dictionary”)比对词集中实体名称, 若二者与词典中名称(或别名、编号等)相同, 即可认定发现了一个实体对象;
(6) 挖掘出所需实体并记录其所在文献的PMID号, 用于后续关联挖掘。
通过上述算法, 将糖尿病相关文献摘要中的基因、药物实体名称进行处理和合并, 最终得到规范化的0-D方体数据。
数据立方体(Data Cube)[16]由维和事实定义, 维是一个单位(或一次研究)想要记录的透视或实体, 常用于商业数据关联挖掘。本文将从PubMed中下载的生物医学文献作为数据仓库, 创新性地提出将生物实体(如: 疾病、基因、药物)作为维, 其中每个维都有与之相关联的表, 该表称之为维表。同时使用support、lift的值作为事实度量标准, 这样即将生物实体关联转变为立方体中维与维之间的关联。
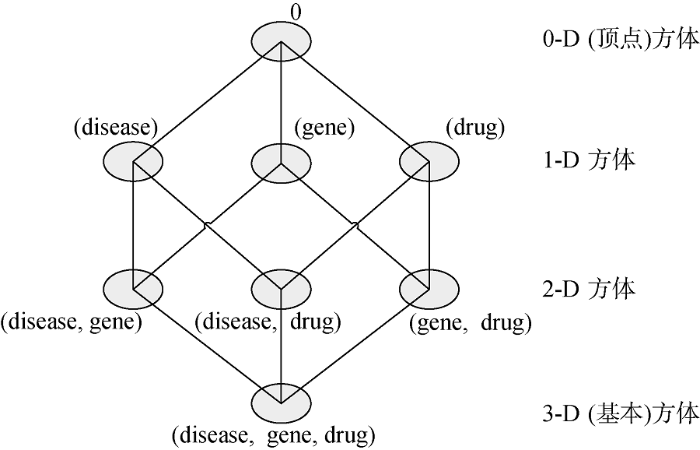
在数据立方体中, 以disease、gene、drug三个属性作为维, 以support和lift的值作为度量, 将该立方体计算的方体或分组总数为8个, 分别为{(disease, gene, drug), (disease, gene), (disease, drug), (gene, drug), (disease), (gene), (drug), ( )}, 其中( )意味着分组为空, 所以实际上有7个分组, 这些分组构成了该数据立方体的方格体, 如图1所示, 其中顶点方体(或0-D方体)表示分组为空的情况, 包含所有可能关联。
(注: 由方体的格组成三维数据立方体, 每一个立方体代表一个不同的分组; 基本方体包含三个维: disease、gene和drug。)
在数据立方体中度量关联时, 需要用到以下术语与度量指标:
(1) 支持度support用于衡量集合内各项出现的频次阈值, 如公式(1)所示。
$\text{support}(A\Rightarrow B)=P(A\bigcup B)=a/N${Invalid MML} (1)
(2) 提升指数lift能够评估一个预测模型是否有效, 体现集合{A}对{B}的重要性, 如公式(2)所示。
$\text{lift}(A\Rightarrow B)=\frac{\text{confidence}(A\Rightarrow B)}{P(A)}=\frac{P(A\bigcup B)}{P(A)P(B)}$ (2)
若值为1, 则A与B无关联; 若值小于1, 则A与B相斥; 若值大于1, 则值越高, A与B之间的关联规则越有价值。本文考虑相关实体可能只是在文献摘要中偶尔或对比提及, 不属于研究内容, 所以设定life阈值为3, 即置信度在99.8%以上或关键值标准偏差是标准正态分布3倍以上, 即认为两者具有强关联性, 如lift>3就是具有强关联性。
本文的数据立方体实际上是一个稀疏冰山立方体(Iceberg Cube)[17], 因此适合使用自底向上构造(Bottom- Up Construction, BUC)算法[18]构建此数据立方体的关联网络, 该算法自顶向下钻, 即从高聚集单元向较低、更细化的单元移动, 详细算法见文献[17-18]。
构建实体关联网络后, 可以发现有些关联(即网络中的边)是生物医学资料中从未报道过的新关联, 也就是关联挖掘的假阳性结果, 但这并不意味着这些结果没有用处, 恰恰相反, 这也是生物实体关联挖掘的主要目的之一, 预测新实体关联[19]。这样通过构建三者关联网络再挖掘出的实体新关联(边), 比以往两两实体预测新关联, 具有更高的可信度, 还可能挖掘出更深层次的新关联。最后使用关联规则将所得预测结果计算并排序, 列出可能性最大的实体新关联, 为生物学研究者设计实验方向提供数据支持。
R语言是一种为统计计算和绘图而生的语言和环境, 包含超过5 000种开源包(如igraph扩展包), 能够较为轻松地构建关联网络[20]。ROC曲线检测算法的准确性适用于二分类情况, 现已广泛应用于医学诊断实验性能的评价[21]。因此, 本文采用R语言实现关联网络, 并用ROC曲线判别算法性能。
从Entrez GENE[22-23]、Gene Ontology[24]、OMIM[25]、DrugBank[26]等数据库中获取并建立基因和药物标准词典, 命名为“Gene_Dictionary”(共计40 172个人类基因词条)和“Drug_Dictionary”(共计1 763种药物词条)词典, 词典包括每个基因(药物)的标准名称、别名、同义词、标准编号等属性。以这两个词典为标准进行命名实体识别。
其次, 以糖尿病为例, 在PubMed中使用“("diabetes mellitus" [MeSH Terms] OR ("diabetes" [All Fields] AND "mellitus" [All Fields]) OR "diabetes mellitus" [All Fields] OR "diabetes" [All Fields] OR "diabetes insipidus" [MeSH Terms] OR ("diabetes" [All Fields] AND "insipidus" [All Fields]) OR "diabetes insipidus" [All Fields]) AND ("2014/08/20" [PDAT] : "2015/08/20" [PDAT])”为检索策略, 获取一年内与糖尿病相关文献共计37 373篇, 并以文本格式保存至本地磁盘。由于本文是对文献的摘要进行实体关联挖掘, 所以剔除其他无用信息(如作者、发表日期等)。
糖尿病分1型糖尿病、2型糖尿病等多种不同病症, 为了深入探讨疾病基因药物之间的关联, 需要对糖尿病进一步分类。在MeSH词表中糖尿病属于营养代谢系统疾病和内分泌系统疾病, 分别存在7种分类, 糖尿病并发症是相关症状的总称, 如表1所示。其中, “Diabetes Mellitus, Type 1”和“Diabetes Mellitus, Type 2”以下简写为“T1DM”和“T2DM”。
表1 糖尿病在MeSH词表中的分类
| 营养性系统疾病下的分类 | 内分泌系统疾病下的分类 | 糖尿病并发症的分类 | |||
|---|---|---|---|---|---|
| 英文名称 | 中文名称 | 英文名称 | 中文名称 | 英文名称 | 中文名称 |
| Diabetes Mellitus, Experimental | 实验性糖尿病 | Diabetes Complications | 糖尿病并发症 | Diabetic Angiopathies | 糖尿病性血管病 |
| Diabetes Mellitus, Type 1 | 1型糖尿病 | Diabetes, Gestational | 妊娠糖尿病 | Diabetic Cardiomyopathies | 糖尿病性心肌病 |
| Diabetes Mellitus, Type 2 | 2型糖尿病 | Diabetes Mellitus, Experimental | 实验性糖尿病 | Diabetic Coma | 糖尿病性昏迷 |
| Diabetes, Gestational | 妊娠糖尿病 | Diabetes Mellitus, Type 1 | 1型糖尿病 | Diabetic Ketoacidosis | 糖尿病性酮症酸中毒 |
| Diabetic Ketoacidosis | 糖尿病酮症酸中毒 | Diabetes Mellitus, Type 2 | 2型糖尿病, | Diabetic Nephropathies | 糖尿病性肾病 |
| Donohue Syndrome | 多诺霍综合症 | Donohue Syndrome | 多诺霍综合症 | Diabetic Neuropathies | 糖尿病性神经病 |
| Prediabetic State | 糖尿病前期 | Prediabetic State | 糖尿病前期 | Fetal Macrosomia | 巨大胎儿(症) |
本文的0-D顶点方体, 即预处理后得到的“(all)词项集”, 是糖尿病数据立方体的顶点, 也是后续研究的数据基础。
综合表1, 去重后得到: 实验性糖尿病、1型糖尿病、糖尿病性血管病、糖尿病性昏迷等共计14种糖尿病相关病症, 由此构建数据立方体中1-D方体(disease)维; 以“Gene_Dictionary”词典为标准, 对糖尿病数据立方体中的0-D顶点立方体进行过滤, 由于可能部分基因在摘要中只是偶尔提及, 为了排除干扰, 设定support阈值为0.1%, 得到ABCC8等23种基因的support值满足大于最小支持度(≥0.1%)的条件, 由此构建1-D方体(gene)维; 以“Drug_Dictionary”词典为标准, 对糖尿病数据立方体中的0-D顶点方体进行过滤, 设定support阈值为0.1%, 得到三磷酸腺苷等24种药物的support值满足大于最小支持度(≥0.1%)的条件, 由此构建1-D方体(drug)维。
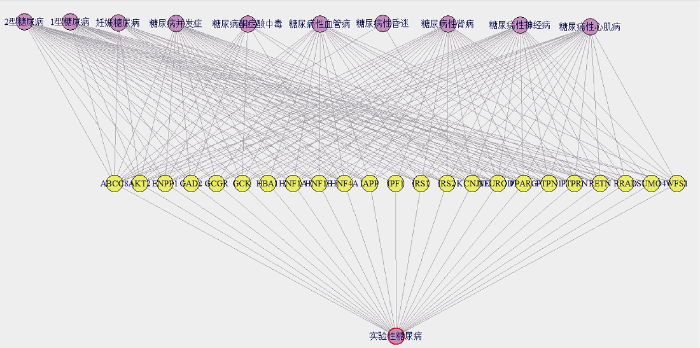
依据前述关联算法, 得到14种糖尿病相关病症和23种基因产生的194种关联, 其中2型糖尿病、糖尿病性神经病、糖尿病性肾病和实验性糖尿病与23种基因均具有关联; 1型糖尿病不与IPF1和SUMO4关联, 与其他21种基因相关; 糖尿病性心肌病不与基因GAD2、IPF1和SUMO4关联; 糖尿病性血管病不与基因DAD2、IPF1、PTPRN和SUMO4相关; 与妊娠糖尿病、糖尿病酮症酸中毒、糖尿病性昏迷相关的基因分别有11、8、2种。由此得到2-D(disease, gene)方体, 并生成糖尿病相关疾病基因关联网络, 如图2所示。
图2中的实验性糖尿病、2型糖尿病、糖尿病性肾病和糖尿病性神经病这4种糖尿病相关病症与23种基因均具有关联。1型糖尿病不与基因IPF1、SUMO4相关, 糖尿病性血管病不与基因GAD2、IPF1、PTPRN、SUMO4相关, 糖尿病并发症不与基因SUMO4、WFS1相关, 糖尿病性心肌病不与基因GAD2、IPF1、SUMO4相关, 但这4种病症与剩下的其他基因具有关联性。多诺霍综合症、糖尿病前期和巨大胎儿(症)与本文得到的23种基因均不具有关联性。
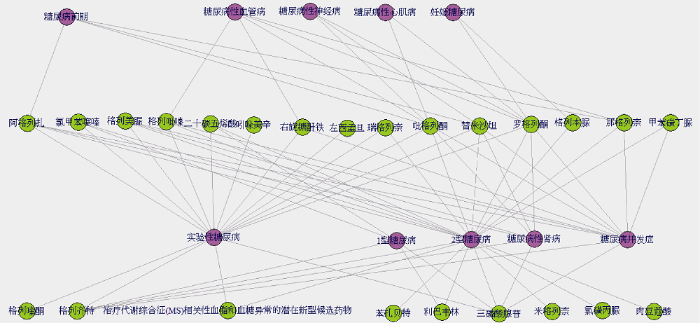
通过关联算法计算, 有10种药物与糖尿病相关病症无关, 分别是: β-D-葡萄糖(Beta-D-Glucose)、糖类多酮类复合化合物19(Compound 19)、布洛芬(Ibuprofen)等; 有4种病症与药物之间不存在关联, 分别是: 糖尿病性昏迷、巨大胎儿(症)、糖尿病酮症酸中毒和多诺霍综合症。最终得到24种药物和11种糖尿病相关病症, 以及它们之间的75种关联, 由此生成2-D(disease, drug)方体, 使用R语言构建该关联网络, 如图3所示。
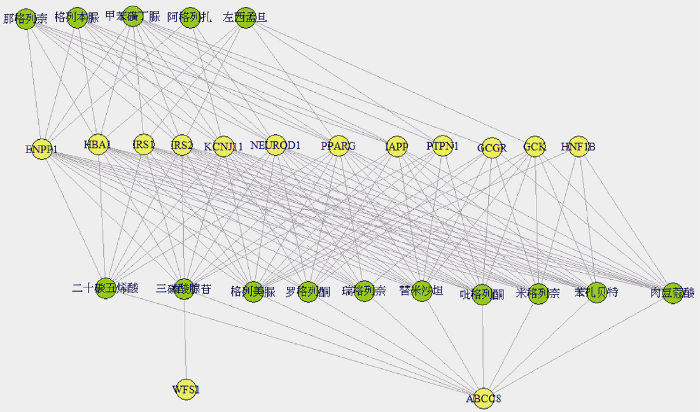
通过关联算法, 得到14种基因与15种药物组成的142种关联, 构成了2-D(gene, drug)方体, 关联网络如图4所示, 得到基因WFS1与药物三磷酸腺苷具有单相关性, 而其他的关联均具有多重性, 即一种基因关联多种药物或一种药物关联多种基因。如: 基因ABCC8对应10种药物, 药物二十碳五烯酸对应8种基因。ATP敏感性钾通道中产生的变体E23K和S1369A可以在基因ABCC8、KCNJ11中找到, 这2个变体可能会对一些药物, 如: 那格列奈等, 在治疗2型糖尿病的过程中产生抑制力[27]。
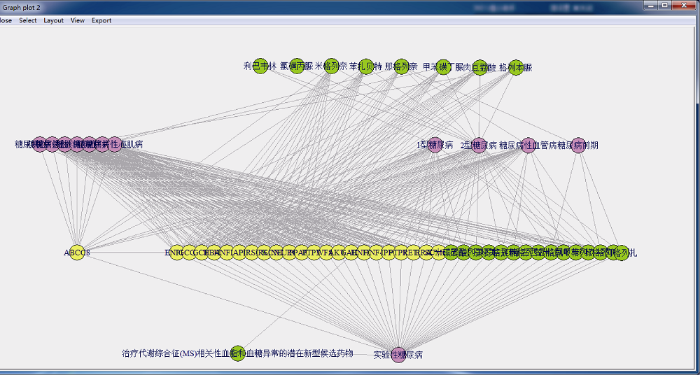
使用BUC算法构建糖尿病基因药物数据立方体, 设定最小lift阈值为3, 去重后, 14种糖尿病病症、23种基因和24种药物之间得到411种关联, 使用R语言构建出糖尿病数据立方体的(disease, gene, drug)3-D基本方体的关联网络, 如图5所示。
同时, 为了深入探讨每种糖尿病病症的疾病基因药物之间的关联, 分别对8种糖尿病症状构建子网模型, 如图6所示。
图6 8种疾病关联子网
(注: a: 2型糖尿病; b: 实验性糖尿病; c: 糖尿病血管病; d: 糖尿病性神经病; e: 糖尿病心肌病; f: 糖尿病肾病; g: 1型糖尿病; h: 妊娠糖尿病)
相关研究发现, 苯扎贝特与2型糖尿病有较大关联, 对于治疗2型糖尿病具有较好的疗效, 有助于血糖调节[28]; 同样, 格列喹酮在实验性糖尿病中的治疗效果, 也有相关文献[29]进行过报道; 2型糖尿病的易感基因KCNJ11部分发病机制也得到验证[30]。
由图5和图6可得, 糖尿病疾病、基因和药物这三者都均有关联性的有318组, 如: (1型糖尿病, ABCC8, 三磷酸腺苷), (2型糖尿病, ABCC8, 苯扎贝特), (实验性糖尿病, ENPP1, 瑞格列奈)等。从疾病角度分析, 有9种糖尿病病症存在三者关联, 如: 1型糖尿病有19组, 32种两两关联; 2型糖尿病有126组, 153种关联等, 其中妊娠糖尿病组数最少, 只有8组15种关联, 与5种基因和2种药物之间存在关联。在糖尿病并发症中, 糖尿病性肾病组数最多, 有60组及80种两两关联, 属于关联网络中的关键节点。
本文将整个关联网络以糖尿病相关病症为标准, 分解出8个子关联网络, 更有助于发现糖尿病相关病症中的候选基因和候选药物, 以及推断疾病、基因药物间的新关联。例如: 对疾病基因2-D方体的关联网络研究发现, 基因ABCC8和KCNJ11与2型糖尿病具有相关性, 这两种基因的变异可以引起新生儿童糖尿病以及家族性持续性高胰岛素低血糖症[31], 但在不同人种中的实验结果存在差异。
部分糖尿病的疾病-基因、疾病-药物和基因-药物之间关联程度排名靠前但尚未报道的实体对如表2所示, 其中基因名参照《英汉人类基因词典》[32]。
表2 预测部分关联程度较高但尚未证实的生物实体间新关联
| Rel | EN 1 | Description 1 | EN 2 | Description 2 |
|---|---|---|---|---|
| Disease-Gene | Diabetic Neuropathies | 糖尿病性神经病 | IPF1 | transcription factor 1 |
| Diabetic Neuropathies | 糖尿病性神经病 | SUMO4 | small ubiquitin-like modifier 4 | |
| Diabetic Nephropathies | 糖尿病性肾病 | IPF1 | transcription factor 1 | |
| Diabetic Nephropathies | 糖尿病性肾病 | SUMO4 | small ubiquitin-like modifier 4 | |
| Disease-Drug | Iron Dextran | 右旋糖酐铁 | Diabetic Angiopathies | 糖尿病性血管病 |
| GFT505 | 治疗代谢综合征(MS)相关性血脂和血糖 异常的潜在新型候选药物 | T2DM | 2型糖尿病 | |
| Telmisartan | 替米沙坦 | Diabetic Neuropathies | 糖尿病性神经病 | |
| Aleglitazar | 阿格列扎 | Diabetic Nephropathies | 糖尿病性肾病 | |
| Gene-Drug | IRS2 | insulin receptor substrate 2 | Icosapent | 二十碳五烯酸 |
| PPARG | peroxisome proliferator-activated receptor gamma | Icosapent | 二十碳五烯酸 | |
| IRS2 | insulin receptor substrate 2 | Levosimendan | 左西孟旦 | |
| GCK | glucokinase (hexokinase 4) | Levosimendan | 左西孟旦 | |
| ENPP1 | ectonucleotide pyrophosphatase/ phosphodiesterase 1 | Myristic Acid | 肉豆蔻酸 |
表2中尚未证实的成对关联, 可为研究人员提供新的研究思路, 例如: 目前尚无文献报道基因SUMO4与糖尿病性神经病、糖尿病性肾病之间是否存在关联, 不过, 文献[33]指出, 1型糖尿病患者中的SUMO4基因多态性M55V与糖尿病性视网膜病变的患病率降低有关, 认为通过SUMO4蛋白质转译后的修改可能导致某些糖尿病并发症的发展, 它们之间存在关联的可能性较大。有报道[34]称某位患者体内的基因ABCC8的34号外显子突变, 导致新生儿肾病, 但是由于该患者开始时被误诊为1型糖尿病, 从而错过了最佳治疗时间, 最终发展为肾病晚期, 这也间接证明了基因ABCC8与糖尿病性肾病可能具有相关性。
对本文得到糖尿病的疾病-基因、疾病-药物和基因-药物之间的所有关联结果进行准确性验证, 关联验证标准[14]如下:
(1) 真阳性(TP): 有已知且确定的直接关联或共现次数大于等于3, 例如: 2型糖尿病与基因ABCC8[35];
(2) 假阳性(FP): 无直接关联且共现次数小于3。
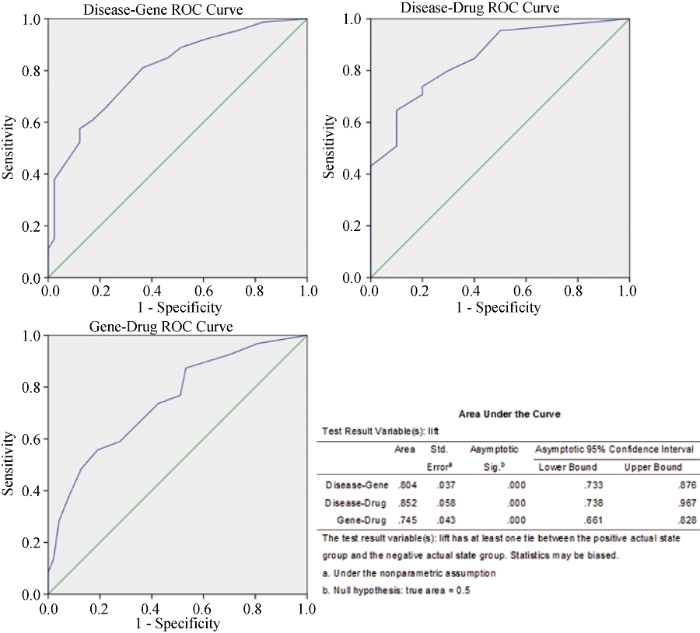
在SPSS20环境下使用ROC曲线判断算法性能, 如图7所示。ROC曲线下的面积分别为0.804、0.815和0.745, 关联准确度中等偏上, 相应的标准误分别为0.037、0.076和0.043, P值均为0.000, 95%置信区间分别为(0.733, 0.876)、(0.666, 0.964)和(0.661, 0.828)。
与其他关联挖掘算法[36-37]类似, 本文也得到一些假阳性(预测性)结果, 这也是生物医学实体关联挖掘的目标之一: 提出预测性的研究假设, 帮助科研人员设计相关实验方向[38]。
空间数据立方体概念建模需要定义两种元数据: 一是来自多种数据源、可维护的、集成的、具有模型数据结构的仓库元数据; 二是可以满足决策者分析需求的、集成的仓库元数据[39]。本文定义识别得到的实体(如疾病、基因、药物)为第一种元数据, 而文献资料和词典则定义为第二种元数据, 因此, 得到一种基于网络的多维数据集模型。
此外, 本文并没有检索到一些糖尿病常用药物: 如胰岛素(Insulin)和二甲双胍(Metformin)等, 原因如下: 本算法检索时使用的药物词典是FDA公布的Drug_Bank数据库, 其中胰岛素有9种名称: (Insulin Regular)、(Insulin Glargine)、(Insulin Lispro)、(Insulin, Porcine)、(Inhaled insulin)、(Insulin Aspart)、(Insulin Detemir)、(Insulin Glulisine)和(Insulin, Isophane), 在文献摘要中完全匹配的检索结果均为零; 而二甲双胍在这一年内的糖尿病相关文献摘要中, 只检索到10篇(support值约为0.026%), 小于设定的support阈值(=0.1%)。
本研究扩展了网络模式分析疾病-药物-基因关联, 网络中的节点代表生物医学实体存储在RDF三元组(即疾病、药物、基因), 边表示生物医学实体间的关联(如“谓词”关系)。为简单起见, 关联均设为单向关联, 丢弃了边的方向和类型, 即只要两节点间有关联, 便认为这两个节点间有边。这样简化疾病药物基因的关联网络中的网络模式有两点作用: 基本可以代表疾病基因药物之间的相互关系; 反映了一个可以有效实现特定功能的框架。
本文创新在于: 在生物实体关联挖掘研究领域, 提出一种基于数据立方体的新方法, 挖掘实体关联, 并结合关联规则对实体关联程度进行分析排序; 以疾病-基因-药物这三种不同生物实体为研究对象, 挖掘新关联, 而CoPub[14]挖掘的是基因-疾病、药物-疾病的关联, PubGene[15]仅挖掘基因-基因间的关联, Sun等[36]挖掘药物-药物间的关联; 使用ROC曲线验证本文算法得到曲线下面积分别为0.804、0.815和0.745, 优于同类算法(如: CoPub和PubGene), 因此本文算法性能更高。下一步工作是在更大规模数据中评估本算法的性能, 确保推广效果。
魏星: 研究方法设计与实现, 论文撰写、修改以及最终版本修订;
胡德华: 提出总体研究思路, 论文修改;
易敏寒: 医学用语修订;
朱启贞, 朱文婕: 算法实现与验证。
所有作者声明不存在利益冲突关系。
支撑数据由作者自存储, E-mail: weixing911119@163.com。
[1] 魏星. DM_pubmed_result.txt. 数据立方体0-D维.
[2] 魏星. 1-D维.xlsx. 数据立方体1-D维.
[3] 魏星. DM_DiseaseGeneDrug.xlsx. 生物实体关联数据.
[4] 魏星. DISEASE-GENE.sav. 疾病-基因ROC曲线验证.
[5] 魏星. DISEASE-Drug.sav. 疾病-药物ROC曲线验证.
[6] 魏星. GENE-Drug.sav. 基因-药物ROC曲线验证.
| [1] |
Computational Tools for Prioritizing Candidate Genes: Boosting Disease Gene Discovery [J].https://doi.org/10.1038/nrg3253 URL PMID: 22751426 [本文引用: 1] 摘要
Abstract At different stages of any research project, molecular biologists need to choose - often somewhat arbitrarily, even after careful statistical data analysis - which genes or proteins to investigate further experimentally and which to leave out because of limited resources. Computational methods that integrate complex, heterogeneous data sets - such as expression data, sequence information, functional annotation and the biomedical literature - allow prioritizing genes for future study in a more informed way. Such methods can substantially increase the yield of downstream studies and are becoming invaluable to researchers.
|
| [2] |
RelEx——Relation Extraction Using Dependency Parse Trees [J].https://doi.org/10.1093/bioinformatics/btl616 URL PMID: 17142812 [本文引用: 1] 摘要
Motivation: The discovery of regulatory pathways, signal cascades, metabolic processes or disease models requires knowledge on individual relations like e.g. physical or regulatory interactions between genes and proteins. Most interactions mentioned in the free text of biomedical publications are not yet contained in structured databases. Results: We developed RelEx, an approach for relation extraction from free text. It is based on natural language preprocessing producing dependency parse trees and applying a small number of simple rules to these trees. We applied RelEx on a comprehensive set of one million MEDLINE abstracts dealing with gene and protein relations and extracted -150 000 relations with an estimated perfomance of both 80% precision and 80% recall. Availability: The used natural language preprocessing tools are free for use for academic research. Test sets and relation term lists are available from our website(http://www.bioifiImu.de/publications/RElEx/).
|
| [3] |
A Novel Feature-Based Approach to Extract Drug-Drug Interactions from Biomedical Text [J].https://doi.org/10.1093/bioinformatics/btu557 URL PMID: 25143286 [本文引用: 1] 摘要
Knowledge of drug-drug interactions (DDIs) is crucial for health-care professionals to avoid adverse effects when co-administering drugs to patients. As most newly discovered DDIs are made available through scientific publications, automatic DDI extraction is highly relevant.We propose a novel feature-based approach to extract DDIs from text. Our approach consists of three steps. First, we apply text preprocessing to convert input sentences from a given dataset into structured representations. Second, we map each candidate DDI pair from that dataset into a suitable syntactic structure. Based on that, a novel set of features is used to generate feature vectors for these candidate DDI pairs. Third, the obtained feature vectors are used to train a support vector machine (SVM) classifier. When evaluated on two DDI extraction challenge test datasets from 2011 and 2013, our system achieves F-scores of 71.1% and 83.5%, respectively, outperforming any state-of-the-art DDI extraction system.The source code is available for academic use at http://www.biosemantics.org/uploads/DDI.zip.
|
| [4] |
Large-scale Extraction of Accurate Drug-Disease Treatment Pairs from Biomedical Literature for Drug Repurposing [J].https://doi.org/10.1186/1471-2105-14-1 URL PMID: 23323762 [本文引用: 1] 摘要
Abstract BACKGROUND: Recent studies of transcription activator-like (TAL) effector domains fused to nucleases (TALENs) demonstrate enormous potential for genome editing. Effective design of TALENs requires a combination of selecting appropriate genetic features, finding pairs of binding sites based on a consensus sequence, and, in some cases, identifying endogenous restriction sites for downstream molecular genetic applications. RESULTS: We present the web-based program Mojo Hand for designing TAL and TALEN constructs for genome editing applications (http://www.talendesign.org). We describe the algorithm and its implementation. The features of Mojo Hand include (1) automatic download of genomic data from the National Center for Biotechnology Information, (2) analysis of any DNA sequence to reveal pairs of binding sites based on a user-defined template, (3) selection of restriction-enzyme recognition sites in the spacer between the TAL monomer binding sites including options for the selection of restriction enzyme suppliers, and (4) output files designed for subsequent TALEN construction using the Golden Gate assembly method. CONCLUSIONS: Mojo Hand enables the rapid identification of TAL binding sites for use in TALEN design. The assembly of TALEN constructs, is also simplified by using the TAL-site prediction program in conjunction with a spreadsheet management aid of reagent concentrations and TALEN formulation. Mojo Hand enables scientists to more rapidly deploy TALENs for genome editing applications.
|
| [5] |
Data Cube. A Relational Aggregation Operator Generalizing Group-By, Cross-Tab, and Sub-Total [J].https://doi.org/10.1023/A:1009726021843 URL [本文引用: 1] 摘要
Data analysis applications typically aggregate data across manydimensions looking for anomalies or unusual patterns. The SQL aggregatefunctions and the GROUP BY operator produce zero-dimensional orone-dimensional aggregates. Applications need the N-dimensionalgeneralization of these operators. This paper defines that operator, calledthe data cube or simply cube. The cube operator generalizes the histogram,cross-tabulation, roll-up,drill-down, and sub-total constructs found in most report writers.The novelty is that cubes are relations. Consequently, the cubeoperator can be imbedded in more complex non-procedural dataanalysis programs. The cube operator treats each of the Naggregation attributes as a dimension of N-space. The aggregate ofa particular set of attribute values is a point in this space. Theset of points forms an N-dimensional cube. Super-aggregates arecomputed by aggregating the N-cube to lower dimensional spaces.This paper (1) explains the cube and roll-up operators, (2) showshow they fit in SQL, (3) explains how users can define new aggregatefunctions for cubes, and (4) discusses efficient techniques tocompute the cube. Many of these features are being added to the SQLStandard.
|
| [6] |
Computational Approaches to Disease-Gene Prediction: Rationale, Classification and Successes [J].https://doi.org/10.1111/j.1742-4658.2012.08471.x URL PMID: 22221742 [本文引用: 1] 摘要
The identification of genes involved in human hereditary diseases often requires the time-consuming and expensive examination of a great number of possible candidate genes, since genome-wide techniques such as linkage analysis and association studies frequently select many hundreds of ‘positional’ candidates. Even considering the positive impact of next-generation sequencing technologies, the prioritization of candidate genes may be an important step for disease-gene identification. In this paper we develop a basic classification scheme for computational approaches to disease-gene prediction and apply it to exhaustively review bioinformatics tools that have been developed for this purpose, focusing on conceptual aspects rather than technical detail and performance. Finally, we discuss some past successes obtained by computational approaches to illustrate their beneficial contribution to medical research.
|
| [7] |
The Human Disease Network [J].https://doi.org/10.1073/pnas.0701361104 URL [本文引用: 1] |
| [8] |
Network-Based Elucidation of Human Disease Similarities Reveals Common Functional Modules Enriched for Pluripotent Drug Targets [J].https://doi.org/10.1371/journal.pcbi.1000662 URL [本文引用: 1] |
| [9] |
Network Systems Biology for Drug Discovery [J].https://doi.org/10.1038/clpt.2010.91 URL PMID: 20520604 [本文引用: 1] 摘要
Systems biology provides a platform for integrating multiple components and interactions underlying cell, organ, and organism processes in health and disease. Beyond traditional approaches focused on individual molecules or pathways, bioinformatic network analysis of high-throughput data sets offers an opportunity for integration of biological complexity and multilevel connectivity. Emerging applications in rational drug discovery range from targeting and modeling disease-corrupted networks to screening chemical or ligand libraries to identification/validation of drug-target interactions for improved efficacy and safety.
|
| [10] |
The Connectivity Map: Using Gene-Expression Signatures to Connect Small Molecules, Genes, and Disease [J].https://doi.org/10.1126/science.1132939 URL PMID: 17008526 [本文引用: 1] 摘要
To pursue a systematic approach to the discovery of functional connections among diseases, genetic perturbation, and drug action, we have created the first installment of a reference collection of gene-expression profiles from cultured human cells treated with bioactive small molecules, together with pattern-matching software to mine these data. We demonstrate that this "Connectivity Map" resource can be used to find connections among small molecules sharing a mechanism of action, chemicals and physiological processes, and diseases and drugs. These results indicate the feasibility of the approach and suggest the value of a large-scale community Connectivity Map project.
|
| [11] |
Inductive Matrix Completion for Predicting Gene-Disease Associations [J].https://doi.org/10.1093/bioinformatics/btu269 URL PMID: 4058925 [本文引用: 1] 摘要
Most existing methods for predicting causal disease genes rely on specific type of evidence, and are therefore limited in terms of applicability. More often than not, the type of evidence available for diseases varies-for example, we may know linked genes, keywords associated with the disease obtained by mining text, or co-occurrence of disease symptoms in patients. Similarly, the type of evidence available for genes varies-for example, specific microarray probes convey information only for certain sets of genes. In this article, we apply a novel matrix-completion method called Inductive Matrix Completion to the problem of predicting gene-disease associations; it combines multiple types of evidence (features) for diseases and genes to learn latent factors that explain the observed gene-disease associations. We construct features from different biological sources such as microarray expression data and disease-related textual data. A crucial advantage of the method is that it is inductive; it can be applied to diseases not seen at training time, unlike traditional matrix-completion approaches and network-based inference methods that are transductive.Comparison with state-of-the-art methods on diseases from the Online Mendelian Inheritance in Man (OMIM) database shows that the proposed approach is substantially better-it has close to one-in-four chance of recovering a true association in the top 100 predictions, compared to the recently proposed Catapult method (second best) that has <15% chance. We demonstrate that the inductive method is particularly effective for a query disease with no previously known gene associations, and for predicting novel genes, i.e. genes that are previously not linked to diseases. Thus the method is capable of predicting novel genes even for well-characterized diseases. We also validate the novelty of predictions by evaluating the method on recently reported OMIM associations and on associations recently reported in the literature.Source code and datasets can be downloaded from http://bigdata.ices.utexas.edu/project/gene-disease.
|
| [12] |
Efficient Mining of Discriminative Co-clusters from Gene Expression Data [J].https://doi.org/10.1007/s10115-013-0684-0 URL PMID: 4308820 [本文引用: 1] 摘要
Discriminative models are used to analyze the differences between two classes and to identify class-specific patterns. Most of the existing discriminative models depend on using the entire feature spa
|
| [13] |
A Random Set Scoring Model for Prioritization of Disease Candidate Genes Using Protein Complexes and Data-Mining of GeneRIF, OMIM and PubMed Records [J].https://doi.org/10.1186/1471-2105-15-315 URL PMID: 154876224409799996603 [本文引用: 1] 摘要
Abstract BACKGROUND: Prioritizing genetic variants is a challenge because disease susceptibility loci are often located in genes of unknown function or the relationship with the corresponding phenotype is unclear. A global data-mining exercise on the biomedical literature can establish the phenotypic profile of genes with respect to their connection to disease phenotypes. The importance of protein-protein interaction networks in the genetic heterogeneity of common diseases or complex traits is becoming increasingly recognized. Thus, the development of a network-based approach combined with phenotypic profiling would be useful for disease gene prioritization. RESULTS: We developed a random-set scoring model and implemented it to quantify phenotype relevance in a network-based disease gene-prioritization approach. We validated our approach based on different gene phenotypic profiles, which were generated from PubMed abstracts, OMIM, and GeneRIF records. We also investigated the validity of several vocabulary filters and different likelihood thresholds for predicted protein-protein interactions in terms of their effect on the network-based gene-prioritization approach, which relies on text-mining of the phenotype data. Our method demonstrated good precision and sensitivity compared with those of two alternative complex-based prioritization approaches. We then conducted a global ranking of all human genes according to their relevance to a range of human diseases. The resulting accurate ranking of known causal genes supported the reliability of our approach. Moreover, these data suggest many promising novel candidate genes for human disorders that have a complex mode of inheritance. CONCLUSION: We have implemented and validated a network-based approach to prioritize genes for human diseases based on their phenotypic profile. We have devised a powerful and transparent tool to identify and rank candidate genes. Our global gene prioritization provides a unique resource for the biological interpretation of data from genome-wide association studies, and will help in the understanding of how the associated genetic variants influence disease or quantitative phenotypes.
|
| [14] |
Literature Mining for the Discovery of Hidden Connections Between Drugs, Genes and Diseases [J]. |
| [15] |
A Literature Network of Human Genes for High-Throughput Analysis of Gene Expression [J].https://doi.org/10.1038/ng0501-21 URL PMID: 11326270 [本文引用: 2] 摘要
Abstract We have carried out automated extraction of explicit and implicit biomedical knowledge from publicly available gene and text databases to create a gene-to-gene co-citation network for 13,712 named human genes by automated analysis of titles and abstracts in over 10 million MEDLINE records. The associations between genes have been annotated by linking genes to terms from the medical subject heading (MeSH) index and terms from the gene ontology (GO) database. The extracted database and accompanying web tools for gene-expression analysis have collectively been named 'PubGene'. We validated the extracted networks by three large-scale experiments showing that co-occurrence reflects biologically meaningful relationships, thus providing an approach to extract and structure known biology. We validated the applicability of the tools by analyzing two publicly available microarray data sets.
|
| [16] |
DADA: A Data Cube for Dominant Relationship Analysis [C]// |
| [17] |
Computing Iceberg Queries Efficiently [C]// |
| [18] |
Bottom-Up Computation of Sparse and Iceberg CUBEs [C]// |
| [19] |
Recent Advances and Emerging Applications in Text and Data Mining for Biomedical Discovery [J].https://doi.org/10.1093/bib/bbv087 URL PMID: 4719073 [本文引用: 1] 摘要
Precision medicine will revolutionize the way we treat and prevent disease. A major barrier to the implementation of precision medicine that clinicians and translational scientists face is understanding the underlying mechanisms of disease. We are starting to address this challenge through automatic approaches for information extraction, representation and analysis. Recent advances in text and data mining have been applied to a broad spectrum of key biomedical questions in genomics, pharmacogenomics and other fields. We present an overview of the fundamental methods for text and data mining, as well as recent advances and emerging applications toward precision medicine.
|
| [20] |
R: A Language and Environment for Statistical Computing [J].https://doi.org/10.1890/0012-9658(2002)083[3097:CFHIWS]2.0.CO;2 URL [本文引用: 1] 摘要
## ## To cite R in publications use: ## ## R Core Team (2012). R: A language and environment for ## statistical computing. R Foundation for Statistical Computing, ## Vienna, Austria. ISBN 3-900051-07-0, URL ## http://www.R-project.org/. ## ## A BibTeX entry for LaTeX users
|
| [21] |
The Meaning and Use of the Area Under a Receiver Operating Characteristic (ROC) Curve [J].https://doi.org/10.1148/radiology.143.1.7063747 URL PMID: 7063747 [本文引用: 1] 摘要
A representation and interpretation of the area under a receiver operating characteristic (ROC) curve obtained by the "rating" method, or by mathematical predictions based on patient characteristics, is presented. It is shown that in such a setting the area represents the probability that a randomly chosen diseased subject is (correctly) rated or ranked with greater suspicion than a randomly chosen non-diseased subject. Moreover, this probability of a correct ranking is the same quantity that is estimated by the already well-studied nonparametric Wilcoxon statistic. These two relationships are exploited to (a) provide rapid closed-form expressions for the approximate magnitude of the sampling variability, i.e., standard error that one uses to accompany the area under a smoothed ROC curve, (b) guide in determining the size of the sample required to provide a sufficiently reliable estimate of this area, and (c) determine how large sample sizes should be to ensure that one can statistically detect differences in the accuracy of diagnostic techniques.
|
| [22] |
Entrez Gene: Gene-Centered Information at NCBI [J].https://doi.org/10.1093/nar/gki031 URL PMID: 17148475 [本文引用: 1] 摘要
Abstract Entrez Gene (www.ncbi.nlm.nih.gov/entrez/query.fcgi?db=gene) is NCBI's database for gene-specific information. It does not include all known or predicted genes; instead Entrez Gene focuses on the genomes that have been completely sequenced, that have an active research community to contribute gene-specific information, or that are scheduled for intense sequence analysis. The content of Entrez Gene represents the result of curation and automated integration of data from NCBI's Reference Sequence project (RefSeq), from collaborating model organism databases, and from many other databases available from NCBI. Records are assigned unique, stable and tracked integers as identifiers. The content (nomenclature, map location, gene products and their attributes, markers, phenotypes, and links to citations, sequences, variation details, maps, expression, homologs, protein domains and external databases) is updated as new information becomes available. Entrez Gene is a step forward from NCBI's LocusLink, with both a major increase in taxonomic scope and improved access through the many tools associated with NCBI Entrez.
|
| [23] |
NCBI Reference Sequences (RefSeq): A Curated Non-Redundant Sequence Database of Genomes Transcripts and Proteins [J].https://doi.org/10.1093/nar/gki025 URL PMID: 15608248 [本文引用: 1] 摘要
ABSTRACT The National Center for Biotechnology Information (NCBI) Reference Sequence (RefSeq) database (http://www.ncbi.nlm.nih.gov/RefSeq/) provides a non-redundant collection of sequences representing genomic data, transcripts and proteins. Although the goal is to provide a comprehensive dataset representing the complete sequence information for any given species, the database pragmatically includes sequence data that are currently publicly available in the archival databases. The database incorporates data from over 2400 organisms and includes over one million proteins representing significant taxonomic diversity spanning prokaryotes, eukaryotes and viruses. Nucleotide and protein sequences are explicitly linked, and the sequences are linked to other resources including the NCBI Map Viewer and Gene. Sequences are annotated to include coding regions, conserved domains, variation, references, names, database cross-references, and other features using a combined approach of collaboration and other input from the scientific community, automated annotation, propagation from GenBank and curation by NCBI staff.
|
| [24] |
Gene Ontology: Tool for the Unification of Biology [J].https://doi.org/10.1038/75556 URL [本文引用: 1] |
| [25] |
Online Mendelian Inheritance in Man (OMIM), A Knowledgebase of Human Genes and Genetic Disorders [J].https://doi.org/10.1093/nar/gki033 URL PMID: 15608251 [本文引用: 1] 摘要
Online Mendelian Inheritance in Man (OMIM) is a comprehensive, authoritative and timely knowledgebase of human genes and genetic disorders compiled to support human genetics research and education and the practice of clinical genetics. Started by Dr Victor A. McKusick as the definitive reference Mendelian Inheritance in Man, OMIM (http://www.ncbi.nlm.nih.gov/omim/) is now distributed electronically by the National Center for Biotechnology Information, where it is integrated with the Entrez suite of databases. Derived from the biomedical literature, OMIM is written and edited at Johns Hopkins University with input from scientists and physicians around the world. Each OMIM entry has a full-text summary of a genetically determined phenotype and/or gene and has numerous links to other genetic databases such as DNA and protein sequence, PubMed references, general and locus-specific mutation databases, HUGO nomenclature, MapViewer, GeneTests, patient support groups and many others. OMIM is an easy and straightforward portal to the burgeoning information in human genetics.
|
| [26] |
DrugBank 3.0: A Comprehensive Resource for ‘Omics’ Research on Drugs [J].https://doi.org/10.1093/nar/gkq1126 URL PMID: 3013709 [本文引用: 1] 摘要
Abstract DrugBank (http://www.drugbank.ca) is a richly annotated database of drug and drug target information. It contains extensive data on the nomenclature, ontology, chemistry, structure, function, action, pharmacology, pharmacokinetics, metabolism and pharmaceutical properties of both small molecule and large molecule (biotech) drugs. It also contains comprehensive information on the target diseases, proteins, genes and organisms on which these drugs act. First released in 2006, DrugBank has become widely used by pharmacists, medicinal chemists, pharmaceutical researchers, clinicians, educators and the general public. Since its last update in 2008, DrugBank has been greatly expanded through the addition of new drugs, new targets and the inclusion of more than 40 new data fields per drug entry (a 40% increase in data 'depth'). These data field additions include illustrated drug-action pathways, drug transporter data, drug metabolite data, pharmacogenomic data, adverse drug response data, ADMET data, pharmacokinetic data, computed property data and chemical classification data. DrugBank 3.0 also offers expanded database links, improved search tools for drug-drug and food-drug interaction, new resources for querying and viewing drug pathways and hundreds of new drug entries with detailed patent, pricing and manufacturer data. These additions have been complemented by enhancements to the quality and quantity of existing data, particularly with regard to drug target, drug description and drug action data. DrugBank 3.0 represents the result of 2 years of manual annotation work aimed at making the database much more useful for a wide range of 'omics' (i.e. pharmacogenomic, pharmacoproteomic, pharmacometabolomic and even pharmacoeconomic) applications.
|
| [27] |
Pharmacogenomic Analysis of ATP-Sensitive Potassium Channels Coexpressing the Common Type 2 Diabetes Risk Variants E23K and S1369A [J].https://doi.org/10.1097/FPC.0b013e32835001e7 URL PMID: 22209866 [本文引用: 1] 摘要
Abstract OBJECTIVES: The common ATP-sensitive potassium (KATP) channel variants E23K and S1369A, found in the KCNJ11 and ABCC8 genes, respectively, form a haplotype that is associated with an increased risk for type 2 diabetes. Our previous studies showed that KATP channel inhibition by the A-site sulfonylurea gliclazide was increased in the K23/A1369 haplotype. Therefore, we studied the pharmacogenomics of seven clinically used sulfonylureas and glinides to determine their structure-activity relationships in KATP channels containing either the E23/S1369 nonrisk or K23/A1369 risk haplotypes. RESEARCH DESIGN AND METHODS: The patch-clamp technique was used to determine sulfonylurea and glinide inhibition of recombinant human KATP channels containing either the E23/S1369 or the K23/A1369 haplotype. RESULTS: KATP channels containing the K23/A1369 risk haplotype were significantly less sensitive to inhibition by tolbutamide, chlorpropamide, and glimepiride (IC50 values for K23/A1369 vs. E23/S1369=1.15 vs. 0.71 mol/l; 4.19 vs. 3.04 mol/l; 4.38 vs. 2.41 nmol/l, respectively). In contrast, KATP channels containing the K23/A1369 haplotype were significantly more sensitive to inhibition by mitiglinide (IC50=9.73 vs. 28.19 nmol/l for K23/A1369 vs. E23/S1369) and gliclazide. Nateglinide, glipizide, and glibenclamide showed similar inhibitory profiles in KATP channels containing either haplotype. CONCLUSION: Our results demonstrate that the ring-fused pyrrole moiety in several A-site drugs likely underlies the observed inhibitory potency of these drugs on KATP channels containing the K23/A1369 risk haplotype. It may therefore be possible to tailor existing therapy or design novel drugs that display an increased efficacy in type 2 diabetes patients homozygous for these common KATP channel haplotypes.
|
| [28] |
Balanced Pan-PPAR Activator Bezafibrate in Combination with Statin: Comprehensive Lipids Control and Diabetes Prevention? [J].https://doi.org/10.1186/1475-2840-11-140 URL PMID: 3502168 [本文引用: 1] 摘要
All fibrates are peroxisome proliferators-activated receptors (PPARs)-alpha agonists with ability to decrease triglyceride and increase high density lipoprotein- cholesterol (HDL-C). However, bezafibrate has a unique characteristic profile of action since it activates all three PPAR subtypes (alpha, gamma and delta) at comparable doses. Therefore, bezafibrate operates as a pan-agonist for all three PPAR isoforms. Selective PPAR gamma agonists (thiazolidinediones) are used to treat type 2 diabetes mellitus (T2DM). They improve insulin sensitivity by up-regulating adipogenesis, decreasing free fatty acid levels, and reversing insulin resistance. However, selective PPAR gamma agonists also cause water retention, weight gain, peripheral edema, and congestive heart failure. The expression of PPAR beta/ delta in essentially all cell types and tissues (ubiquitous presence) suggests its potential fundamental role in cellular biology. PPAR beta/ delta effects correlated with enhancement of fatty acid oxidation, energy consumption and adaptive thermogenesis. Together, these data implicate PPAR beta/delta in fuel combustion and suggest that pan-PPAR agonists that include a component of PPAR beta/delta activation might offset some of the weight gain issues seen with selective PPAR gamma agonists, as was demonstrated by bezafibrate studies. Suggestively, on the whole body level all PPARs acting as one orchestra and balanced pan-PPAR activation seems as an especially attractive pharmacological goal. Conceptually, combined PPAR gamma and alpha action can target simultaneously insulin resistance and atherogenic dyslipidemia, whereas PPAR beta/delta properties may prevent the development of overweight. Bezafibrate, as all fibrates, significantly reduced plasma triglycerides and increased HDL-C level (but considerably stronger than other major fibrates). Bezafibrate significantly decreased prevalence of small, dense low density lipoproteins particles, remnants, induced atherosclerotic plaque regression in thoracic and abdominal aorta and improved endothelial function. In addition, bezafibrate has important fibrinogen-related properties and anti-inflammatory effects. In clinical trials bezafibrate was highly effective for cardiovascular risk reduction in patients with metabolic syndrome and atherogenic dyslipidemia. The principal differences between bezafibrate and other fibrates are related to effects on glucose level and insulin resistance. Bezafibrate decreases blood glucose level, HbA1C, insulin resistance and reduces the incidence of T2DM compared to placebo or other fibrates. Currently statins are the cornerstone of the treatment and prevention of cardiovascular diseases related to atherosclerosis. However, despite the increasing use of statins as monotherapy for low density lipoprotein- cholesterol (LDL-C) reduction, a significant residual cardiovascular risk is still presented in patients with atherogenic dyslipidemia and insulin resistance, which is typical for T2DM and metabolic syndrome. Recently, concerns were raised regarding the development of diabetes in statin-treated patients. Combined bezafibrate/statin therapy is more effective in achieving a comprehensive lipid control and residual cardiovascular risk reduction. Based on the beneficial effects of pan-PPAR agonist bezafibrate on glucose metabolism and prevention of new-onset diabetes, one could expect a neutralization of the adverse pro-diabetic effect of statins using the strategy of a combined statin/fibrate therapy.
|
| [29] |
Gliquidone Decreases Urinary Protein by Promoting Tubular Reabsorption in Diabetic Goto- Kakizaki Rats [J].https://doi.org/10.1530/JOE-13-0199 URL PMID: 24254365 摘要
Abstract The efficacy of gliquidone for the treatment of diabetic nephropathy was investigated by implanting micro-osmotic pumps containing gliquidone into the abdominal cavities of Goto-Kakizaki (GK) rats with diabetic nephropathy. Blood glucose, 2466h urinary protein, and 2466h urinary albumin levels were measured weekly. After 4 weeks of gliquidone therapy, pathological changes in the glomerular basement membrane (GBM) were examined using an electron microscope. Real-time PCR, western blotting, and immunohistochemistry were employed to detect glomerular expression of receptors for advanced glycation end products (RAGE) (AGER), protein kinase C β (PKCβ), and protein kinase A (PKA) as well as tubular expression of the albumin reabsorption-associated proteins: megalin and cubilin. Human proximal tubular epithelial cells (HK-2 cells) were used to analyze the effects of gliquidone and advanced glycation end products (AGEs) on the expression of megalin and cubilin and on the absorption of albumin. Gliquidone lowered blood glucose, 2466h urinary protein, and 2466h urinary albumin levels in GK rats with diabetic nephropathy. The level of plasma C-peptide increased markedly and GBM and podocyte lesions improved dramatically after gliquidone treatment. Glomerular expression of RAGE and PKCβ decreased after gliquidone treatment, while PKA expression increased. AGEs markedly suppressed the expression of megalin and cubulin and the absorption of albumin in HK-2 cells in vitro, whereas the expression of megalin and cubilin and the absorption of albumin were all increased in these cells after gliquidone treatment. In conclusion, gliquidone treatment effectively reduced urinary protein in GK rats with diabetic nephropathy by improving glomerular lesions and promoting tubular reabsorption.
|
| [30] |
An Isogenic Human ESC Platform for Functional Evaluation of Genome-wide- Association-Study-Identified Diabetes Genes and Drug Discovery [J].https://doi.org/10.1016/j.stem.2016.07.002 URL PMID: 27524441 [本文引用: 1] 摘要
Genome-wide association studies (GWASs) have increased our knowledge of loci associated with arange of human diseases. However, applying such findings to elucidate pathophysiology and promote drug discovery remains challenging. Here, we created isogenic human ESCs (hESCs) with mutations in GWAS-identified susceptibility genes for type 2 diabetes. In pancreatic beta-like cells differentiated from these lines, we found that mutationsinCDKAL1, KCNQ1, and KCNJ11 led to impaired glucose secretion invitro and invivo, coinciding with defective glucose homeostasis. CDKAL1 mutant insulin+ cells were also hypersensitive to glucolipotoxicity. A high-content chemical screen identified a candidate drug that rescued CDKAL1-specific defects invitro and invivo by inhibiting theFOS/JUN pathway. Our approach of a proof-of-principle platform, which uses isogenic hESCs for functional evaluation of GWAS-identified loci and identification of a drug candidate that rescues gene-specific defects, paves the way for precision therapy of metabolic diseases.
|
| [31] |
Beta-cell Hyperexcitability: From Hyperinsulinism to Diabetes [J].https://doi.org/10.1111/j.1463-1326.2007.00778.x URL PMID: 17919182 [本文引用: 1] 摘要
Abstract Nutrient oxidation in beta cells generates a rise in [ATP]:[ADP] ratio. This reduces K(ATP) channel activity, leading to depolarization, activation of voltage-dependent Ca(2+) channels, Ca(2+) entry and insulin secretion. Consistent with this paradigm, loss-of-function mutations in the genes (KCNJ11 and ABCC8) that encode the two subunits (Kir6.2 and SUR1, respectively) of the ATP-sensitive K(+) (K(ATP)) channel underlie hyperinsulinism in humans, a genetic disorder characterized by dysregulated insulin secretion. In mice with genetic suppression of K(ATP) channel subunit expression, partial loss of K(ATP) channel conductance also causes hypersecretion, but unexpectedly, complete loss results in an undersecreting, mildly glucose-intolerant phenotype. When challenged by a high-fat diet, normal mice and mice with reduced K(ATP) channel density respond with hypersecretion, but mice with more significant or complete loss of K(ATP) channels cross over, or progress further, to an undersecreting, diabetic phenotype. It is our contention that in mice, and perhaps in humans, there is an inverse U-shaped response to hyperexcitabilty, leading first to hypersecretion but with further exacerbation to undersecretion and diabetes. The causes of the overcompensation and diabetic susceptibility are poorly understood but may have broader implications for the progression of hyperinsulinism and type 2 diabetes in humans.
|
| [32] |
|
| [33] |
A M55V Polymorphism in the SUMO4 Gene is Associated with a Reduced Prevalence of Diabetic Retinopathy in Patients with Type 1 Diabetes [J].https://doi.org/10.1055/s-2007-985357 URL PMID: 17926234 摘要
Abstract AIMS: We studied the association between a functionally relevant M55V polymorphism in the SUMO4 gene with microvascular diabetic complications in patients with type 1 diabetes. METHODS: 223 patients with type 1 diabetes were studied using polymerase chain reaction and subsequent cleavage by restriction endonucleases for the M55V SUMO4 gene variant. RESULTS: No effect of the polymorphism on diabetic neuropathy or diabetic nephropathy was found, but heterozygous or homozygous patients for the M55V polymorphism in the SUMO4 gene had a markedly reduced prevalence of diabetic retinopathy (odds ratio 0.37, 95%-confidence interval (CI) [0.32;0.43]; p=0.004). Furthermore, a multiple logistic regression model showed an age and diabetes duration independent effect of the M55V polymorphisms on the prevalence of diabetic retinopathy (p=0.03), but not of diabetic neuropathy or nephropathy. CONCLUSIONS: Our data indicate that the M55V polymorphism in the SUMO4 gene is associated with a reduced risk of diabetic retinopathy in type 1 diabetes. Thus, the results of our study suggest that posttranslational modification of proteins via SUMO4 could contribute to the development of certain diabetic complications.
|
| [34] |
Neonatal Diabetes with End-Stage Nephropathy Pancreas Transplantation Decision [J].https://doi.org/10.2337/dc08-0823 URL [本文引用: 1] |
| [35] |
The Common C49620T Polymorphism in the Sulfonylurea Receptor Gene (ABCC8), Pancreatic Beta Cell Function and Long-Term Diabetic Complications in Obese Patients with Long-Lasting Type 2 Diabetes Mellitus [J]. |
| [36] |
Extracting Drug-Drug Interactions from Literature Using a Rich Feature-Based Linear Kernel Approach [J].https://doi.org/10.1016/j.jbi.2015.03.002 URL PMID: 25796456 [本文引用: 2] 摘要
Identifying unknown drug interactions is of great benefit in the early detection of adverse drug reactions. Despite existence of several resources for drug-drug interaction (DDI) information, the wealth of such information is buried in a body of unstructured medical text which is growing exponentially. This calls for developing text mining techniques for identifying DDIs. The state-of-the-art DDI extraction methods use Support Vector Machines (SVMs) with non-linear composite kernels to explore diverse contexts in literature. While computationally less expensive, linear kernel-based systems have not achieved a comparable performance in DDI extraction tasks. In this work, we propose an efficient and scalable system using a linear kernel to identify DDI information. The proposed approach consists of two steps: identifying DDIs and assigning one of four different DDI types to the predicted drug pairs. We demonstrate that when equipped with a rich set of lexical and syntactic features, a linear SVM classifier is able to achieve a competitive performance in detecting DDIs. In addition, the one-against-one strategy proves vital for addressing an imbalance issue in DDI type classification. Applied to the DDIExtraction 2013 corpus, our system achieves an F1 score of 0.670, as compared to 0.651 and 0.609 reported by the top two participating teams in the DDIExtraction 2013 challenge, both based on non-linear kernel methods.
|
| [37] |
Large-scale Automatic Extraction of Side Effects Associated with Targeted Anticancer Drugs from Full-Text Oncological Articles [J].https://doi.org/10.1016/j.jbi.2015.03.009 URL PMID: 25817969 [本文引用: 1] 摘要
We downloaded 13,855 full-text articles from the Journal of Oncology (JCO) published between 1983 and 2013. We developed text classification, relationship extraction, signaling filtering, and signal prioritization algorithms to extract drug–SE pairs from downloaded articles. We extracted a total of 26,264 drug–SE pairs with an average precision of 0.405, a recall of 0.899, and an F 1 score of 0.465. We show that side effect knowledge from JCO articles is largely complementary to that from the US Food and Drug Administration (FDA) drug labels. Through integrative correlation analysis, we show that targeted drug-associated side effects positively correlate with their gene targets and disease indications. In conclusion, this unique database that we built from a large number of high-profile oncological articles could facilitate the development of computational models to understand toxic effects associated with targeted anticancer drugs.
|
| [38] |
Recent Advances and Emerging Applications in Text and Data Mining for Biomedical Discovery [J].https://doi.org/10.1093/bib/bbv087 URL PMID: 4719073 [本文引用: 1] 摘要
Precision medicine will revolutionize the way we treat and prevent disease. A major barrier to the implementation of precision medicine that clinicians and translational scientists face is understanding the underlying mechanisms of disease. We are starting to address this challenge through automatic approaches for information extraction, representation and analysis. Recent advances in text and data mining have been applied to a broad spectrum of key biomedical questions in genomics, pharmacogenomics and other fields. We present an overview of the fundamental methods for text and data mining, as well as recent advances and emerging applications toward precision medicine.
|
| [39] |
Conceptual Model for Spatial Data Cubes: A UML Profile and Its Automatic Implementation [J].https://doi.org/10.1016/j.csi.2014.06.004 URL [本文引用: 1] 摘要
61The paper presents a state of art of conceptual models for Spatial Data Warehouses61The paper details a new UML profile for modeling complex Spatial OLAP applications61The implementation in a commercial CASE tool is presented61Its automatic implementation in a classical ROLAP architecture is shown
|

/
| 〈 |
|
〉 |



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}